Кожа человека является важной анатомической структурой и играет огромную роль в его жизни. Кожные покровы защищают организм от проникновения инфекции, травм, переохлаждений и перегреваний, участвует в регуляции водного и минерального обмена и многое другое. Но при развитии кожных заболеваний страдает не только кожа, но и организм в целом. Особенно тяжело протекает такая патология, как буллезный эпидермолиз. Данное заболевание протекает очень тяжело при отсутствии должного ухода за больным и трудно поддается лечению.
Содержание
- 1 Что это за болезнь
- 2 Причины
- 3 Классификация
- 4 Клиническая картина
- 5 Продолжительность жизни больных
- 6 Диагностика
- 7 Лечение
- 8 Вопрос – ответ
- 9 МКБ-10
- 10 Общие сведения
- 11 Причины буллезного эпидермолиза
- 12 Классификация буллезного эпидермолиза
- 13 Симптомы буллезного эпидермолиза
- 14 Диагностика буллезного эпидермолиза
- 15 Лечение буллезного эпидермолиза
- 16 Прогноз буллезного эпидермолиза
- 17 Что такое буллёзный эпидермолиз?
- 18 Сколько людей болеют буллёзным эпидермолизом?
- 19 Причины буллёзного эпидермолиза
- 20 Классификация буллёзного эпидермолиза
- 21 Проявления буллёзного эпидермолиза
- 22 Простой буллёзный эпидермолиз
- 23 Пограничный буллёзный эпидермолиз
- 24 Дистрофический буллёзный эпидермолиз
- 25 Киндлер-синдром
- 26 Осложнения буллёзного эпидермолиза
- 27 Продолжительность жизни при буллёзном эпидермолизе
- 28 Диагностика буллёзного эпидермолиза
- 29 Принципы лечения буллёзного эпидермолиза
- 30 Заключение
Что это за болезнь
Врожденный буллезный эпидермолиз включает в себя целую группу заболеваний (более 30 форм), которые объединены одним названием. Общий признак этих болезней – появление на коже пузырей при малейшей механической травме. Данную патологию называют еще болезнью бабочки, но не от того, что вырастают крылья у больных детей, а от характерной для них чрезмерной хрупкости и уязвимости кожных покровов (у бабочки легко повредить крылья). В механизме возникновения пузырей лежит сдвиг между верхними и нижними слоями кожи. Назвать болезнь данным термином предложил немецкий врач – дерматолог Генрих Кёбнер. Группа данных заболеваний генетически детерминирована и может наследоваться по аутосомно-рециссивному и по аутосомно-доминантному типу, доказано, что мутации затрагивают более 10 генов.
Эпидемиология
 Болезнь буллезный эпидермолиз распространена на территории 70 (всего 85) субъектов РФ. Частота ее составляет 0 – 19, 73 случаев на 1000000 населения. В России заболеваемость буллезным эпидермолизом достигает 1:50 тыс. – 1:300 тыс. населения.
Болезнь буллезный эпидермолиз распространена на территории 70 (всего 85) субъектов РФ. Частота ее составляет 0 – 19, 73 случаев на 1000000 населения. В России заболеваемость буллезным эпидермолизом достигает 1:50 тыс. – 1:300 тыс. населения.
Исследователи патологии выявили, что во многих странах мира в структуре болезни преобладает простой буллезный эпидермолиз, а в некоторых странах – дистрофический буллезный эпидермолиз. Половые различия для данной патологии не характерны. Отмечено, что среди зарегистрированных пациентов преобладают дети и подростки, что связано с высокой смертностью таких больных.
Причины
В норме слои кожи человека скрепляются между собой различными по составу веществами. В основе развития данной патологии лежит дефект крепления между эпидермисом (поверхностный сой кожи) и дермой (более глубокий слой). Развитие заболевания обусловлено генными мутациями в тех генах, которые отвечают за синтез структурных белков кожи – обеспечивающих механизм крепления между разными ее слоями. На сегодняшний день обнаружено больше 1000 мутаций в 15 генах кожных структурных белков, которые приводят к возникновению различных форм буллезного эпидермолиза. Мутации вызывают нарушение образования белков: полное отсутствие белка, производство неполноценного белка, который не способен «склеивать» слои кожи, синтез белка с нарушенным строением, из-за чего протеазы (ферменты, разрушающие белки) получают легкий доступ к белку. Таким образом становится понятно, что причинами патологии являются генные нарушения и болезнь передается по наследству.
При простой форме заболевания мутации присутствуют в генах: KRT5, KRT14, РКР -1, PLEC, ITGA6, ITGB4. При этом образуются дефектные белки в поверхностном слое кожи (в кератиноцитах): десмоплакин, кератин 5 и 14, плектин и другие, на которые воздействуют протеазы, которые выделяются при механическом воздействии на кожу, что и приводит к образованию пузырей.
Мутации в генах LAMB3, LAMA3, COL7A1 и ряде других вызывают дефект в образовании белков базальной мембраны (нижний слой эпидермиса): ламинин 332, коллаген 17, интегрин и приводят к развитию пограничной формы эпидермиса. Данная форма помимо образования кожных пузырей характеризуется чрезмерной ломкостью кожи и более тяжелым течением.
Дистрофическая форма болезни развивается при мутациях в гене COL7A1, что отвечает за синтез киндлина и коллагена 17 и располагается в соединительнотканных волокнах кожи. Снижение образования данных белков вызывает не только легкое образование пузырей и эрозий, но и приводит к поражению других органов (слизистая дыхательной системы и ЖКТ, контрактуры суставов). Дистрофическая форма болезни протекает тяжело, с образованием грубых рубцов на коже, на которых часто возникают злокачественные образования.
Классификация
На сегодняшний день известно множество разновидностей заболевания и предприняты попытки классифицировать их на определенные типы. Существует множество классификаций, что заставляет путаться в них даже исследователей буллезного эпидермолиза. Самая современная классификация данной патологии, используемая дерматологами, включает 4 основных формы (типа) буллезного эпидермолиза, каждый из которых делится на подтипы:
- Простой буллезный эпидермолиз
К этому типу заболевания относится 12 подтипов. Уровень, на котором образуется пузырь – интраэпидермальный слой. Самыми распространенными подтипами выступают: синдром Вебера-Коккейна, синдром Кёбнера, синдром Доулинг-Меары. Наследование данного типа происходит и аутосомно-доминантно и аутосомно-рецессивно. Пузыри образуются внутриэпидермально и субэпидермально – поражены белки эпидермиса.
- Пограничный буллезный эпидермолиз
Включает 2 подтипа, в одном из которых имеется 6 самостоятельных форм. Уровень образования пузыря – светлая пластинка базальной мембраны эпидермиса. Самой тяжелой формой данного типа выступает подтип Герлитца, смертность при котором очень высока. Из-за образования пузырей на уровне светлой пластинки данная форма получила название «пограничный» эпидермолиз.
- Дистрофический буллезный эпидермолиз
Делится на 2 подтипа, в зависимости от механизма наследования (доминантный или рецессивный). Доминантный встречается чаще. В рецессивный подтип дистрофического эпидермолиза входит несколько форм, одна из которых самая тяжелая – подтип Аллопо-Сименса. Уровень образования пузырей – сосочковый слой дермы, что вызывает образование длительно заживающих эрозий и появление грубых шрамов.
Эту форму называют еще смешанным эпидермолизом, она считается редкой и малоизученной. Особенность данного типа – формирование пузырей и в эпидермисе, и в дерме, и на уровне светлой пластинки.
Клиническая картина
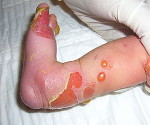
Ведущим клиническим признаком заболевания при любой его форме является возникновение пузырей в ответ на незначительную механическую травму. Подтипы простого эпидермолиза характеризуются образованием полушаровидных, напряженных и заполненных жидкостью пузырей, которые формируются в местах трения/травмирования. Пограничная и дистрофическая формы сопровождаются образованием вялых, со складками покрышкой, которая провисает под весом жидкости. Такие пузыри легко вскрываются и образуются не только в районах травмирования, но и в зонах растяжения (складки паха и подмышечные, в области шеи). При любом типе патологии могут формироваться множественные пузыри и эрозии, которые захватывают большую площадь кожи. Эрозии отличаются длительным заживлением при любой разновидности болезни.
Простая форма болезни
Локализация пузырей на стопах и кистях рук, а при тяжелом течении патологии поражается вся кожа больного. Если болезнь протекает легко, то пузыри заживают, не оставляя рубцов. Простая форма делится на 2 больших подтипа: локализованный и генерализованный.
Локализованный простой БЭ или синдром Вебера-Коккейна характеризуется образованием пузырей на ладонях и стопах, а обострение болезни приходится на летний период, когда повышается риск травматизации кожи и усиливается потливость. Чем старше становится ребенок, тем меньше у него образуется пузырей.
Генерализованный простой БЭ или синдром Доулинг-Меары отличается тяжелым течением и характеризуется формированием пузырей у новорожденных сразу после рождения. Прогрессирование болезни отмечается до года, затем процесс стабилизируется и со временем отмечается улучшение вплоть до прекращения возникновения пузырей либо их редкого образования. Часто такие пузыри заполнены гноем и покрыты слоистыми желтоватыми корочками. После того, как пузыри исчезают в местах их локализации отмечается стойкая пигментация.
К дополнительным признакам простого генерализованного БЭ относятся:
- запоры;
- анальные трещины;
- гиперкератоз;
- повреждение слизистых;
- затрудненность дыхания;
- дистрофия ногтей и сращение пальцев;
- патология сердца;
- пищеводный рефлюкс;
- образование миль (белых узелков);
- признаки анемии (бледность и слабость, головокружения);
- базальноклеточная форма рака кожи (у взрослых).
Дистрофическая форма болезни
Дистрофическая форма может наследоваться рецессивно или доминантно. Это самая тяжелая подгруппа БЭ, так как повреждается глубокий слой дермы, то заживление пузырей происходит с формированием грубых рубцов. Почти всегда при дистрофической форме болезни поражаются слизистые. Характерным и наиболее тяжелым подтипом дистрофической формы является синдром Аллопо-Сименса. Проявления заболевания видны уже с самого рождения, а многие новорожденные рождаются с полным отсутствием эпидермиса на конечностях. Особенно тяжело патология протекает в первые годы. Отмечается медленная эпителизация эрозий, вплоть до нескольких месяцев. Характерно, что чем старше ребенок, тем хуже процесс заживления. Образование грубых рубцов приводит к формированию контрактур и сращению пальцев, что в дальнейшем блокирует рост и развитие кистей/стоп. Подобные рубцовые изменения приводят к трудностям самообслуживания и передвижения. Поражение слизистых с образованием эрозий во тру заканчивается рубцеванием и формированием микростомии (сужение ротового отверстия), короткой уздечки языка, дефектом речи. Рубцевание эрозий в глотке/пищеводе ведет к образованию непроходимости и сужению пищевода, трудностям глотания. Заживление эрозий в прямой кишке нарушает дефекацию. Также больные подвержены кариесу и у них нередко имеются аномалии расположения зубов, вплоть до полной из потери.
Пограничная форма болезни
Наиболее тяжелый подтип – это синдром Герлитца, для которого характерны формирование и быстрое распространение вялых и множественных пузырей, которые почти сразу вскрываются. Избыточное разрастание грануляционной ткани ведет к образованию грануляций вокруг ротовой полости и ногтей, что возникают в первые дни жизни младенца. Также ребенка сопровождает аплазия кожи (отсутствие части кожи на волосистом покрове головы), дефект ногтей и охриплость голоса, проблемы с зубами. Заживление эрозий также сопровождается образованием грубых шрамов. Другие симптомы (некожные): задержка роста и умственного развития ребенка, нарушенность дыхания, анемия, проблемы с легкими (пневмония) и пищеварительным трактом (гастроэнтерит) и развитие сепсиса. Летальность при синдроме Герлитца очень высокая, ребенок погибает от дыхательной недостаточности (образование пузыря на слизистой дыхательных путей), сепсиса или дистрофии.
Синдром Киндлера
Данная форма характеризуется возникновением распространенных пузырных образований уже к рождению, светочувствительностью и истонченной, мятой кожей. Некожные симптомы проявляются в виде острого колита, эзофагита, сужения мочеиспускательного канала, выворотом века. Зубы остаются в порядке.
Продолжительность жизни больных
Сколько живут дети-бабочки? Ответить на данный вопрос однозначно затруднительно. Продолжительность жизни больных с буллезным эпидермолизом прямо пропорциональна форме заболевания, степени повреждения строения генов, глубины повреждения кожи и общего состояния ребенка. К сожалению, до зрелого возраста такие больные доживают редко, и в первую очередь это связано с уходом за ними, что требует множество усилий, терпения и мужества. Дети с тяжелыми генерализованными формами болезни живут всего несколько лет и погибают еще в дошкольном возрасте, например, синдром Герлитца или Аллопо-Сименса. Легкие формы, особенно простого БЭ могут со временем войти в стойкую ремиссию, а образование пузырей связано лишь с травматизацией кожи.
Диагностика
Диагноз данного заболевания ставится на основании сбора данных анамнеза и жалоб, общего осмотра пациента и подтверждается лабораторными исследованиями. Для лабораторного анализа необходимо провести гистологическое исследование участка кожи, взятого из места повреждения, где находится свежий пузырь. Гистологический анализ позволяет выявить субэпидермальную полость, но бессилен в определении типа буллезного эпидермолиза. С этой целью проводится исследование с помощью метода непрямой иммунофлюоресценции (НРИФ) или трансмиссионной электронной микроскопии:
- Биоптат кожи, который исследуют при помощи НРИФ, позволяет установить присутствие, снижение или отсутствие синтеза белков кожи. Кроме того, НРИФ помогает определить уровень повреждения кожи и образование пузыря (внутриэпидермальный уровень, внутри светлой пластинки базальной мембраны или под плотной пластинкой). В диагностике имеет значение определение недостающего белка и уровня формирования пузырного образования. Чтобы провести НРИФ, получают биоптат кожи с пузыря, образовавшегося в течение 24 и менее часов. Участок кожи берется на границе здорового места и свежего пузырного образования.
- Методом электронной микроскопии устанавливают подтип заболевания. Выявляются ультраструктурные изменения в биоптате кожи и оценивается состояние клеточных и внеклеточных образований.
Также для диагностики буллезного эпидермолиза привлекаются смежные специалисты (терапевт, гастроэнтеролог, гематолог, кардиолог, хирург, стоматолог, окулист, ЛОР-врач, онколог).
Дифференциальная диагностика
Буллезный эпидермолиз у новорожденных дифференцируют с такими заболеваниями, как:
- пузырчатка новорожденных;
- эксфолиативный дерматит;
- герпес новорожденных;
- буллезная врожденная эритродермия.
В более старшем возрасте заболевание дифференцируют со следующими патологиями:
- герпетиформный дерматит;
- буллезный пемфигоид;
- экссудативная эритема;
- эпидермальный некролиз.
Лечение
Лечение буллезного эпидермолиза задача непростая и преследует следующие цели:
- обработка пузырей и язв;
- предупреждение новых высыпаний;
- ликвидация или уменьшение интенсивности некожных симптомов;
- улучшение качества жизни пациента.
Специфической терапии при данном заболевании не разработано. Применяются симптоматические методы лечения. Больного следует осматривать дважды в день на предмет новых высыпаний. Пузырьковые образования сначала обрабатывают антисептиком, затем вскрывают, используя стерильные ножницы, скальпель или иглу. Пузырь прокалывается в двух местах параллельно коже. Покрышку пузырька снимать не следует, нужно лишь выпустить его содержимое и наложить асептическую повязку с антисептиком (нитрофурал, хлорид метилтиония и прочие). Для перевязки очага поражения используют первичные повязки из коллагеновых пористых материалов и вторичные фиксирующие. Снимают повязки, обработав их очистителем для кожи (спрей).
Если больного беспокоит зуд, назначают антигистамины и глюкокортикоиды. При значительных болях – ненаркотические анальгетики (ибупрофен, кеторол). Если ребенок глотает с трудом, лекарства перорально выписываются в жидком виде (суспензия) или вводятся парентерально. В случае присоединения вторичной инфекции назначаются антибиотики. Также показана обработка неповрежденной кожи мазями с витамином А и косметическими увлажняющими кремами дважды в день. Это повышает защитные свойства кожи. При развитии осложнений к лечению привлекаются смежные специалисты.
Уход за больным
Режим для больного ребенка должен быть охранительным. То есть запрещаются любые физические нагрузки, которые повышают потоотделение, резкие движения и травмоопасные ситуации.
Больные нуждаются в особой диете. Пища должна быть протертой, не холодной и не горячей, полужидкой и без приправ и специй. Диета должна быть богата белками, углеводами и жирами, а также витаминами и минералами – организму требуется строительные вещества для заживления эрозий. Также больной должен потреблять много жидкости (раневые участки кожи теряют тканевую влагу).
Особые требования предъявляются к одежде и обуви. Одежда больного ребенка должна быть свободной, не натирать и не стягивать кожу. Следует отдавать предпочтение одежде из натуральных материалов. Дома желательно одевать ребенка в мягкую (из байки, фланели) пижаму, а на ноги носки (тапки не желательны). Одежда должна быть многослойной, таким образом она сохраняет тепло и не допускает скопления пота. Нижнее белье и первый слой одежды надевать швами наружу. Не допускаются ремешки и пояса, тугие резинки на плавках и пижаме.
Обувь должна быть изготовлена из натуральной, но мягкой кожи, с минимумом швов и отсутствием декоративных деталей. Желательно приобретать обувь на липучках, избегая обувь с молниями. Обувь для ребенка должна быть свободной и легко надеваться, даже с повязками на ногах.
Вопрос – ответ
Нет, особых противопоказаний не существует. Не делают прививки лишь при плохом самочувствии больного (осложнение кожных высыпаний). Также не выполняются пробы Манту.
К сожалению, болезнь неизлечима, так как заложена на генном уровне. Но в медицине описаны редчайшие случаи обратного мозаицизма – восстановление работы организма, в частности, белков кожи и возникновение новых изменений в структуре генов. Но утешить родителей больного ребенка может то, что чем старше становится больной, тем легче протекает болезнь.
Да, в России существует и в настоящее время активно пренатальная диагностика генетических заболеваний, в том числе и буллезного эпидермолиза. С этой целью на сроках 10 – 14 недель выполняется хорионцентез и определяется наличие генетического дефекта у плода. Если он имеется, беременность рекомендуют прервать. Пренатальная диагностика показана семейным парам, у которых уже есть больной малыш или при подтвержденном заболевании одного из родителей.
Да, инвалидность определяется при любой форме заболевания и назначается пенсия.
Буллезный эпидермолиз – группа наследственных заболеваний, которые характеризуются легкой ранимостью кожи, отсюда второе название этих патологий — «механобуллезная болезнь». Основным симптомом служит развитие на поверхности кожных покровов пузырей с серозным содержимым, после чего на их месте возникают долго незаживающие эрозии. Диагностика различных типов буллезного эпидермолиза осуществляется при помощи иммуногистологических и генетических методик, а также на основании данных осмотра пациента и изучения его наследственного анамнеза. Специфического лечения не существует, однако правильная и комплексная симптоматическая терапия может в ряде случаев значительно улучшать состояние больного.
МКБ-10
Общие сведения
Буллезный эпидермолиз – это гетерогенная группа наследственных заболеваний кожи, которые характеризуются образованием пузырей и эрозий в ответ на незначительное механическое воздействие. Впервые данный термин был использован в 1886 году немецким врачом-дерматологом Генрихом Кёбнером, дальнейшие исследования продемонстрировали, что существует множество разновидностей этой патологии. Генетические исследования буллезного эпидермолиза показали, что он может наследоваться как аутосомно-рецессивно, так и аутосомно-доминантно, с ним ассоциированы мутации более чем 10 генов. Существенные различия имеются и в клиническом течении разных типов этого заболевания, встречаемость колеблется в пределах 1:30000-1:1000000.
Патогенез нарушений при буллезном эпидермолизе долгое время оставался малоизученным. Прорыв в этом направлении произошел с внедрением в медицинскую практику электронной микроскопии, которая помогла визуализировать ультраструктуру пораженных тканей кожи. Следующий важный шаг в изучении буллезного эпидермолиза был совершен с открытием иммуногистологических исследований (иммунофлуоресценция). В настоящее время именно эти методики играют важнейшую роль в диагностике данных заболеваний, уступая по точности лишь генетическому анализу. Ввиду того, что методы изучения буллезного эпидермолиза постоянно совершенствовались, претерпевала изменения и классификация форм этой группы заболеваний.
Причины буллезного эпидермолиза
Этиология буллезного эпидермолиза неодинакова у разных типов заболевания, что в некоторых случаях достаточно сильно осложняет диагностику. Простой буллезный эпидермолиз обусловлен мутациями генов KRT5 и KRT14, однако, по данным врачей-генетиков, нарушением структуры этих генов объясняется только 75% случаев заболевания этого типа. При этом в кожных покровах, предположительно, нарушается равновесие в системе «ферменты-ингибиторы», и некоторые белки становятся объектом атаки. При простом буллезном эпидермолизе это могут быть протеины базальной мембраны (альфа6-бета4-интегрин) и белки десмосом базального слоя эпидермиса – десмоплакин, плакофиллин-1. В результате при механическом воздействии происходит выделение ферментов, которые разрушают указанные белки, тем самым провоцируя цитолиз и разрушение структуры эпидермиса, приводя к образованию пузырей.
Причиной развития другой формы патологии – пограничного буллезного эпидермолиза – являются мутации в генах LAMB3, LAMA3 и некоторых других. Большинство из этих мутации наследуется по аутосомно-рецессивному механизму, объектом атаки разбалансированной ферментной системы становятся такие протеины, как коллаген 17-го типа и ламинин-332. Эти белки участвуют в поддержании нормальной структуры нижних слоев эпидермиса, поэтому их повреждение приводит к характерным клиническим симптомам пограничного буллезного эпидермолиза. Помимо легкого образования пузырей и эрозий он характеризуется также повышенной ломкостью кожных покровов и более тяжелым течением.
Дистрофический тип буллезного эпидермолиза обусловлен мутациями в гене COL7A1, которые могут наследоваться как по аутосомно-доминантному, так и аутосомно-рецессивному механизмам. Белком-мишенью при этом выступает коллаген 7-го типа, который отвечает за стабильность структуры других соединительнотканных волокон кожи. Уменьшение количества этого протеина в тканях кожных покровов приводит к легкому развитию высыпаний, эрозий и пузырей, а также нередко сопровождается нарушениями других органов. В частности, дистрофический буллезный эпидермолиз часто приводит к развитию контрактуры суставов, поражение захватывает слизистые оболочки органов дыхательной и пищеварительной систем. На рубцах, которые остаются после заживления эрозий, нередко возникают злокачественные опухоли.
В целом, общий патогенез буллезного эпидермолиза можно свести к нарушению активности некоторых ферментов в тканях кожи. В результате этого разрушаются определенные ключевые структурные белки эпидермиса, дермы или базальной мембраны, что нарушает связи между клетками и приводит к образованию пузырей при механическом воздействии даже незначительной силы. Типы буллезного эпидермолиза отличаются один от другого локализацией пузырьков, видом мутации, что привела к этому заболеванию, и разновидностью белка, который стал объектом атаки ферментов.
Классификация буллезного эпидермолиза
В настоящий момент существуют десятки разновидностей буллезного эпидермолиза, которые достаточно трудно классифицировать в определенные группы. Проблема осложняется еще и тем, что почти за полтора века изучения данной патологии предпринимались неоднократные попытки разделить ее на определенные типы, используя самые современные на тот момент данные. В конечном итоге это привело к некоторой путанице, даже в научной литературе можно найти самые разнообразные варианты разделения буллезного эпидермолиза на разновидности. Наиболее современная классификация этого состояния в дерматологии включает в себя четыре типа заболевания, которые, в свою очередь, делятся на ряд подтипов:
- Простой буллезный эпидермолиз – имеет 12 подтипов, наиболее распространенными из которых являются синдромы Вебера-Коккейна, Кёбнера, Доулинга-Меары. Может наследоваться как аутосомно-доминантно, так и рецессивно, встречаемость составляет 1:100000. Простой буллезный эпидермолиз характеризуется образованием внутриэпидермальных или, реже, субэпидермальных пузырей, так как при этом заболевании поражаются белки эпидермиса.
- Пограничный буллезный эпидермолиз – делится на 2 подтипа, один из которых имеет еще 6 самостоятельных клинических форм. Наиболее тяжелой формой этого заболевания является подтип Херлитца, имеющий крайне высокую смертность. Встречаемость пограничного буллезного эпидермолиза составляет около 1:500000, образование пузырей при нем происходит на уровне светлой пластинки, что и дало ему название «пограничный».
- Дистрофический буллезный эпидермолиз – имеет два подтипа, которые делятся по механизму наследования этой патологии (доминантный и рецессивный подтипы). При этом встречаемость доминантного варианта несколько выше (3:1000000 против 1:500000 у рецессивной формы дистрофического буллезного эпидермолиза). Рецессивная разновидность также имеет несколько клинических форм, наиболее тяжелой из которых является подтип Аллопо-Сименса. При этом варианте заболевания у больных возникают глубокие эрозии, оставляющие после себя шрамы, возможны контрактуры суставов, поражение слизистых оболочек. Образование пузырей при этом происходит в сосочковом слое дермы, что и обуславливает появление шрамов и длительное заживление эрозий.
- Синдром Киндлера, или смешанный буллезный эпидермолиз, является одной из наиболее редких и малоизученных форм данной патологии. Особенностью, которая позволила выделить эту форму в отдельный тип, является образование пузырей во всех слоях кожи – эпидермисе, у светлой пластинке, в дерме. В настоящий момент определен только белок, выступающий в качестве мишени ферментов при смешанном буллезном эпидермолизе – киндлин-1.
Такой тип разделения всех клинических форм буллезного эпидермолиза является в настоящее время общепринятым. Но даже в пределах одного типа наблюдается большое разнообразие клинических симптомов заболевания, что осложняет диагностику и нередко влияет на прогноз патологии. Поэтому на сегодняшний день не прекращаются поиски более структурированной и приемлемой классификации буллезного эпидермолиза.
Симптомы буллезного эпидермолиза
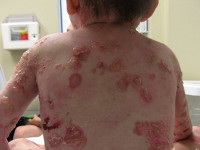
Проявления буллезного эпидермолиза разных типов объединяет одно – развитие пузырей и эрозий в ответ на механическое воздействие на кожу. Различается лишь степень выраженности этих изменений, локализация, время существования и результаты заживления. При локализованной форме простого буллезного эпидермолиза (подтип Вебера-Коккейна) поражения располагаются только на определенном участке тела (руки, стопы). В младенческом возрасте возможна более широкая площадь появления пузырей, но с возрастом их выраженность уменьшается. Напротив, генерализованный подтип Доулинга-Меары характеризуется развитием мелких везикулярных высыпаний на значительной площади тела. Такой тип буллезного эпидермолиза возникает с самого раннего детства и может стать причиной смерти ребенка, итогом разрешения пузырьков может быть гиперкератоз, нарушения пигментации кожи, иногда возникает поражение слизистых.
Пограничная форма буллезного эпидермолиза протекает намного более тяжело, особенно так называемый летальный подтип Херлитца. При этом наблюдается повышенная ломкость кожных покровов, образование большого количества пузырьков, эрозий, на лице и спине часто возникают симметричные грануляции. Поражаются и слизистые оболочки рта, обнаруживается гипоплазия эмали и обусловленный ею тяжелый кариес. Столь тяжелое течение пограничного буллезного эпидермолиза часто становится причиной летального исхода в первые годы жизни. У выживших больных во взрослом возрасте формируются контрактуры суставов, поражение почек, потеря ногтей. Более легкая атрофическая форма пограничного буллезного эпидермолиза также характеризуется обширными высыпаниями, после разрешения которых формируются атрофические участки и рубцы. Также она часто приводит к дистрофии ногтей и рубцовой алопеции.
Дистрофический буллезный эпидермолиз практически всегда является генерализованным и поражает обширные участки тела. Доминантный вариант заболевания в целом отличается более доброкачественным течением, образование пузырей и их разрешение происходит медленно, однако большинство больных в конце концов теряют ногти на руках. После заживления эрозий на поверхности кожи формируются заметные рубцы. Рецессивный вариант дистрофического буллезного эпидермолиза, особенно его тяжелый генерализованный подтип, протекает намного тяжелее: помимо высыпаний у больных часто регистрируются псевдосиндактилии, обширные шрамы, потеря ногтей. Возникает поражение костей скелета, на месте заживших шрамов с годами может развиваться плоскоклеточный рак. Проблемой является еще и высокая устойчивость подтипа Аллопо-Сименса к терапевтическим мероприятиям.
Осложнения любого типа буллезного эпидермолиза сводятся к риску развития шока (при обширных поражениях), присоединения вторичной инфекции и спровоцированного ею сепсиса, обезвоживания больных. В большинстве случаев терапевтические процедуры производят только с целью недопущения этих состояний. Вероятность развития осложнений тем выше, чем большую область тела занимают патологические очаги и чем деструктивнее их характер (напряженные пузыри, эрозии, язвы).
Диагностика буллезного эпидермолиза
В настоящее время диагностика буллезного эпидермолиза осуществляется путем осмотра кожных покровов пациента, с помощью проведения иммуногистологических исследований и генетических анализов, в некоторых случаях производят изучение наследственного анамнеза. При осмотре кожных покровов специалист также может произвести диагностические тесты – механически воздействовать на кожу пациента и спустя время оценить результаты. Развитие на этом участке характерных для буллезного эпидермолиза пузырей или эрозий говорит в пользу наличия данного заболевания. На следующих этапах диагностики производят более точное определение формы патологии.
Иммунофлуоресцентный анализ при буллезном эпидермолизе осуществляется при помощи моно- и поликлональных антител, имеющих сродство к основным белкам эпидермиса, светлой пластинки и верхних слоев дермы. Это позволяет оценить количество того или иного белка, что, в свою очередь, говорит о ферментной активности тканей. Уменьшение количества того или иного белка свидетельствует о его низком выделении или же ускоренном разрушении. Снижение концентрации ключевых протеинов на определенных участках позволяет определить уровень развития пузырей на самом раннем этапе, что уже помогает с высокой долей вероятности определить тип буллезного эпидермолиза. Точку в диагностике этого состояния ставит генетический анализ методом прямого секвенирования генов, которые ассоциированы с тем или иным типом заболевания. Такой многостадийный подход к диагностике буллезного эпидермолиза обеспечивает высокую точность.
Значительно упростить диагностику этого заболевания позволяет изучение наследственного анамнеза пациента, по которому можно выявить его кровных родственников с такой же проблемой. Кроме того, если у кого-то из родных имеется буллезный эпидермолиз, имеет смысл производить пренатальную генетическую диагностику, что позволит выявить наличие данной патологии на ранних этапах развития плода. Дифференциальную диагностику осуществляют с истинной пузырчаткой, некоторыми формами буллезного пемфигоида, приобретенным буллезным эпидермолизом (который является не наследственным, а аутоиммунным заболеванием).
Лечение буллезного эпидермолиза
Специфического лечения этого заболевания не существует, все терапевтические процедуры сводятся к предупреждению развития осложнений и уменьшению выраженности пузырьков и эрозий. В случае тяжелых форм буллезного эпидермолиза назначают преднизолон. Из наружных терапевтических манипуляций производят асептическое вскрытие пузырьков, обработку их крышки антисептиками, накладывают гелиомициновую мазь. Наложение повязок нужно производить крайне осторожно, так как давление бинтов может спровоцировать появление новых пузырей. При наличии осложнений (шока, сепсиса) проводят симптоматическое лечение противошоковыми препаратами и антибиотиками. С профилактической целью можно производить облучение кожных покровов ультрафиолетовыми лучами.
Современная генетика и ряд других областей медицины продолжают широкие исследования буллезного эпидермолиза с целью поиска более эффективных методик лечения. Среди основных технологий и методов наиболее перспективными считаются способы с использованием стволовых клеток, белковая и генная терапии. Однако пока ни один из методов не вышел за рамки экспериментов на животных, поэтому буллезный эпидермолиз в настоящее время является неизлечимым заболеванием.
Прогноз буллезного эпидермолиза
Прогноз буллезного эпидермолиза чаще всего неопределенный, так как зависит от множества факторов и обстоятельств – типа заболевания, наличия или отсутствия у больного сопутствующих нарушений, его образа жизни. Например, локальный подтип простого эпидермолиза чаще всего имеет доброкачественное течение и редко создает угрозу жизни пациенту. Тогда как подтип Аллопо-Сименса имеет очень высокую смертность – как и от кожных проявлений, так и по причине отдаленных осложнений, таких как поражения почек и органов ЖКТ, а также развития плоскоклеточного рака кожи. Больные с такой проблемой должны бережно относиться к своей коже, не забывать про антисептическую обработку эрозий и других поражений, избегать занятий травмирующими видами спорта и иной деятельностью такого рода.
Буллёзный эпидермолиз — это редкая и страшная болезнь, при которой поражаются кожные покровы, а иногда и слизистые оболочки.
По всему миру детишек, страдающих этим недугом, принято называть «бабочками». Сравнение не случайно, ведь их кожа тонка и очень хрупкая, подобно крылу насекомого.
Любые неосторожные прикосновения причиняют малышу боль и оставляют раны. Дети — бабочки страдают не только от своего диагноза, но и от отсутствия знаний у персонала больниц. Первые травмы ребёнок получает ещё в родильном доме. Иногда они даже несовместимы с жизнью. У буллёзного эпидермолиза множество форм, но все они неизлечимы.
![]()
![]()
Что такое буллёзный эпидермолиз?
Буллёзный эпидермолиз включает в себя огромное количество болезней кожи, передающихся по наследству и имеющих общий признак – пузыри разных размеров и разного количества. Они образуются на кожном покрове, либо слизистых оболочках вследствие любого, иногда минимального воздействия. Любое трение, прикосновение, смена температуры и влажности воздуха способны вызвать обострение процесса.
Недуг способен проявиться в разном возрасте. Ребёнок заболевает ещё в утробе матери. Чаще всего он либо уже рождается с признаками болезни, либо они проявляются в ближайшее время после появления на свет. Во всех случаях правильнее называть болезнь врождённый буллёзный эпидермолиз.
Сколько людей болеют буллёзным эпидермолизом?
Чаще всего недуг регистрируется у детишек возраста 1 — 5 лет. Данные Национального регистра больных буллёзным эпидермолизом, который ведётся в Соединённых Штатах Америки, говорят о том, что недугом страдает 1 из 50000 новорождённых. За 16 лет его существования в США выявлены 3300 человек с данной болезнью.
В Европе буллёзным эпидермолизом страдает 1 из 30000 новорождённых малышей. В Японии наименьшая распространённость болезни, она выявляется у 7,8 из 1 млн рождённых детей.
Причины буллёзного эпидермолиза
Причина этой тяжёлой болезни – мутации в генах, которые ответственны за синтез структурных белков кожи. В результате её клетки теряют прочные соединения между собой. Малейшие воздействия извне способствуют повреждениям кожного покрова.
Кожа человека состоит из нескольких слоёв. Верхний – эпидермис представлен клетками кератиноцитами. Они постоянно делятся и по мере своего роста продвигаются из нижележащих слоёв к верхнему – роговому слою, обеспечивая обновление кожного покрова и его защиту. Друг с другом кератиноциты соединяются специальными мостиками – десмосомами, из которых выступают белковые нити – тонофибриллы. Клетки нижних слоёв эпидермиса они соединены ещё и белком ламинином.
![]()
За эпидермисом следует слой, называемый дерма. Он включает в себя коллагеновые, эластические и ретикулярные волокна, которые пронизаны многочисленными кровеносными и лимфатическими сосудами, нервами, потовыми и сальными железами, волосяными фолликулами. Дерма содержит клетки фибробласты. Именно они производят все волокна, находящиеся в составе слоя.
В основе разных клинических форм буллёзного эпидермолиза лежат структурные нарушения кожи, когда, в результате мутации, происходит рост и созревание повреждённых кератиноцитов, отсутствие или уменьшение удерживающих и крепящих белковых нитей. Возможен и дефицит определённых белков, участвующих в строении кожи: коллагенов, кератинов, ламинина и других.
В результате ослабевают связи между слоями кожи и её клетками и при малейшем воздействии извне возникают повреждения с образованием пузырей.
Классификация буллёзного эпидермолиза
Со времен развития технологий, которые позволяют определять строение кожного покрова на микроскопических уровнях и определить её мельчайшие структуры, буллёзный эпидермолиз был разделён на 3 группы. Позднее была выделена ещё одна группа заболевания.
В современном медицинском сообществе классификация заболевания состоит 4 основных групп и 6 подгрупп. В подгруппах собраны разные клинические формы болезни. Они различаются по типу наследования, микроскопическим изменениям в коже, клиническим проявлениям, степени тяжести, прогнозу.
Итак, буллёзный эпидермолиз может быть простым, пограничным, дистрофическим. Отдельной группой выделяется киндлер-синдром. Простой буллёзный эпидермолиз включает в себя супрабазальный и базальный. Формами пограничного буллёзного эпидермолиза могут быть локализованная и генерализованная. Дистрофический буллёзный эпидермолиз бывает доминантный и рециссивный. Киндлер-синдром на подгруппы не делится.
Проявления буллёзного эпидермолиза
Пузыри, эрозии разных размеров на коже и слизистых оболочках – главный признак буллёзного эпидермолиза. Они появляются вследствие уменьшения устойчивости кожи к разным воздействиям из внешней среды. Часто такое происходит при изменении температуры окружающего воздуха, воздействия давления и трения. Из-за того, что кожный покров имеет необычное строение, появляются пузыри, затем – эрозии. Их заживление при некоторых видах буллёзного эпидермолиза может проходить с образованием грубых рубцов.
Простой буллёзный эпидермолиз
Малыши, страдающие простым буллёзным эпидермолизом рождаются с пузырями на коже, либо они появляются в первые месяцы их жизни. Пузыри можно увидеть на кистях, стопах, локтях, коленях, голенях, волосистой части головы. В полости рта их мало или нет совсем. Пузыри и эрозии отличаются безболезненностью и быстрым заживлением.
Ногти не изменяются. Если происходит их отслоение, они обязательно восстанавливаются. Зубки у таких деток здоровы. Пузыри заживают не оставляя следов. С ростом ребёнка они будут образовываться всё реже.
![]()
Локализованный простой буллёзный эпидермолиз кистей и стоп относится к базальному простому буллёзному эпидермолизу. Он проявляется с началом самостоятельной ходьбы. Болезнь может начаться и у подростков, когда начинается ношение тесной обуви и травмируются стопы. Они поражаются чаще всего. Поражение других участков тела встречается реже. Их количество бывает разным, от незначительных высыпаний, до огромных, ограничивающих обычную жизнь.
Генерализованный простой буллёзный эпидермолиз — это тоже одна из клинических форм базального буллёзного эпидермолиза. У малышей-грудничков поражаются затылок, спина, локти. По мере роста и взросления ребёнка пузыри появляются на кистях, стопах, местах, подверженных трениям.
Пузырей много. Они расположены группами, которые формируют очаги самых разных причудливых очертаний. Поражаются слизистые оболочки, легко сходят ногти. Часто меняется пигментация кожи, появляется гипергидроз и гиперкератоз ладоней и подошв. Важно — после повреждений кожи не остаётся рубцов.
Пограничный буллёзный эпидермолиз
При пограничном буллёзном эпидермолизе изменения касаются базальной мембраны эпидермиса. Он подразделяется на генерализованный и локализованный. Общим их признаком является появление пузырей на любом участке тела с обширными поражениями. Также характерны изменения зубной эмали, она истончается, появляются точечные углубления на поверхности зуба. Они часто подвергаются кариесу. Пограничный буллёзный эпидермолиз протекает тяжелее, чем простой.
Генерализованный тяжёлый пограничный буллёзный эпидермолиз ещё называется «летальный». Это смертельное заболевание, исходами которого являются грубое обезображивание и инвалидность. Малыш рождается с пузырями, либо возрастом их проявления служит период новорождённости. Часто пузыри локализуются в периоральной области.
Ими покрыты волосистая часть головы, ноги, промежность, грудная клетка ребёнка. На кистях и стопах они появляются редко, в отличие от других типов буллёзного эпидермолиза. Исключение составляют конечные фаланги пальцев, на которых располагаются ногти. Сами же пластины разрушаются, отслаиваются и навсегда утрачиваются. Часто высыпаниями поражаются все слизистые оболочки.
Генерализованный средне – тяжёлый пограничный буллёзный эпидермолиз отличается от предыдущего варианта более лёгким течением. Пузыри также могут обнаруживаться у новорождённого ребёнка, но при их заживлении рубцов не образуется. Отличительная черта данного клинического варианта – очаговое выпадение волос и выраженная атрофия кожи волосистой части головы. Растут и развиваются такие малыши в соответствии с возрастом. Болезнь на это никак не влияет.
Дистрофический буллёзный эпидермолиз
Данная группа буллёзного эпидермолиза классифицируется на две подгруппы в зависимости от типа наследования: аутосомно-доминантного или аутосомно-рецессивного. Это значит, что генетическая мутация чётко передаётся по наследству. В первом случае для развития болезни достаточно лишь одного мутантного гена, который имеется у отца или матери ребёнка. Во втором случае оба родителя должны быть носителями дефектного гена, но сами они будут здоровы. А вот малыш родится уже с тяжёлым недугом.
Рецессивный дистрофический буллёзный эпидермолиз чаще других форм болезни приводит к тяжёлой инвалидности, хотя тяжесть его может быть не такой сильной. Пузыри могут быть расположены на кистях, стопах, локтях и коленях, либо распространяются по всему телу. Аналогичные изменения появляются на слизистых оболочках. Вследствие образования грубых рубцов малыш становится инвалидом.
Киндлер-синдром
Дети с киндлер-синдромом появляются на свет с пузырями на коже и слизистых оболочках. В старшем возрасте появляются светочувствительность, пигментации на коже, образуется рубцовая ткань, отслаиваются ногти. Характерно поражение желудочно-кишечного тракта и мочевых путей.
Осложнения буллёзного эпидермолиза
Развитие осложнений чаще развиваются у детишек, страдающих пограничным и дистрофическим буллёзным эпидермолизом. Болезни этих групп протекают в самых агрессивных формах.
Обширные язвы и эрозии на коже со временем приводят к тому, что она замещается грубой рубцовой тканью. Она отличается плохой чувствительностью, слабой растяжимостью, отсутствием сальных и потовых желёз. Функции кожи утрачиваются, кроме того, рубцы представляют собой и косметический дефект, уродующий внешность.
![]()
Рубцовое замещение кожи век приводит к ограничению их движения. Малыш не сможет полноценно открыть или закрыть глаз, вследствие чего орган зрения может остаться без защиты. Изменится и форма глаза вплоть до полного его закрытия. В результате присоединяются конъюнктивиты, блефариты и другие воспалительные заболевания глаза. Ребёнок может и вовсе лишиться зрения.
Грубая рубцовая ткань в области суставов приводит к ограничениям движений в них. Ребёнок не сможет полноценно согнуть и разогнуть сустав, так как рубец не сможет полноценно растянуться, как обычная кожа. Сустав чаще остаётся в одном и том же положении, развивается его контрактура – тугоподвижность.
Эрозии и мокнутия в области пальцев приводят к тому, что рубцовая ткань образуется со срастанием пальцев. В таком случае кисти ребёнка навсегда теряют функцию захвата.
Эрозии на слизистых оболочках также могут заживать с образованием рубцовой ткани, приводя к сужениям – стриктурам пищевода, дыхательных и мочевых путей, кишечника. Ребёнок не может полноценно глотать пищу, говорить, дышать, мочиться, следовательно, будет худеть вплоть до кахексии, часто болеть воспалительными заболеваниями лёгких, почек. Всасывание пищи в кишечнике будет нарушено. Это ещё больше усугубит состояние малыша.
У людей, страдающих буллёзным эпидермолизом, высок риск развития плоскоклеточного рака кожи.
Продолжительность жизни при буллёзном эпидермолизе
Продолжительность жизни «детей-бабочек» зависит от формы болезни. Самый благоприятный прогноз у малышей, страдающих простым буллёзным эпидермолизом. Они могут даже полноценно жить и учится. Есть вариант, что с возрастом болезнь вообще отступит.
При других формах болезни, в большинстве случаев, развивается тяжёлая инвалидизация. Но у малышей остаётся шанс прожить долгую жизнь при правильном наблюдении и уходе.
Диагностика буллёзного эпидермолиза
Диагноз буллёзного эпидермолиза не всегда может быть поставлен только на основании жалоб. Чтобы подтвердить или опровергнуть его, нужна сложная лабораторная диагностика в крупных клиниках. Необходим также генетический анализ.
В обязательном порядке проводится биопсия кожи, обязательно из свежего пузыря. Кусочек кожи сразу же должен быть подвержен заморозке в жидком азоте, либо замочен в физиологическом растворе. Для длительного хранения фрагмент кожи помещается в специальную транспортную среду. Биообразец исследуется под специальными мощными микроскопами с обработкой контрастными веществами. Можно определить аномалии в структуре кожи, и выявить недостаток определённых белковых компонентов.
Принципы лечения буллёзного эпидермолиза
В современном мире буллёзный эпидермолиз считается неизлечимым недугом. Действия по лечению и профилактике должны предотвращать травматизацию кожи. Для этого кожа «детей-бабочек» нуждается в подборе правильного ухода. Для комфорта маленьких пациентов важно устраненить зуд и болевые ощущения.
Чтобы предотвратить опасные для жизни осложнения, нужно своевременно бороться с инфицированием кожи. Если развиваются осложнения со стороны органов пищеварения и суставов, нужно вовремя скорректировать их. Родители «детей-бабочек» должны быть обучены уходу за ними в кратчайшие сроки. Для него используются специальные мягкие пластыри, повязки, мази с серебром, антисептики.
![]()
Обычно крупные пузыри аккуратно вскрываются. На образовавшуюся эрозию наносится необходимое средство. Потом рана закрывается повязкой. Пользуются специализированными атравматическими бинтами, которые содержат сорбент, антисептик, регенерирующие и противомикробные средства.
Сверху накладывается вторая повязка. Благодаря ей лечебная повязка хорошо зафиксируется и будет плотно сидеть на месте. Фиксирующая повязка не должна сильно давить на кожу и завязываться на плотный узел, чтобы ещё больше не травмировать кожу. Для профилактики осложнений нужны отдельные перевязки каждого пальца ребёнка. Конечность при бинтовании не должна быть согнута или разогнута. Перевязка малыша с тяжёлыми клиническими вариантами буллёзного эпидермолиза может занимать 1 — 2 часа.
Малыш, страдающий буллёзным эпидермолизом, должен наблюдаться у многих специалистов и регулярно осматриваться дерматологом, педиатром, гастроэнтерологом, отоларингологом, хирургом. По необходимости нужны консультации торакального и пластического хирурга.
В некоторых случаях нужно введение антибиотика в уколах. Ребёнок должен получать с пищей все необходимые питательные вещества и витамины. Детям назначаются специализированные высокобелковые смеси. Если из-за осложнений малыш не может нормально глотать пищу, возможно установление гастростомы.
Заключение
Буллёзный эпидермолиз – это тяжёлое и, к сожалению, пока неизлечимое заболевание. Все мероприятия по лечению носят только паллиативный характер. Но при правильной и своевременно начатой терапии можно значительно облегчить страдания малыша и дать ему шанс на долгую жизнь.